Regulatory aspects to the development of intra-vaginal rings
When a pharmaceutical product or, more specifically, a drug-device-combination product (DDC) is being developed for multiple markets, such as the US and EU, one should comply with the regulations set forth for these territories. This may be challenging as, although there is an overlap in regulations, there are differences in requirements and deliverables. This article reflects on the general concept of DDCs and intra-vaginal rings. It presents the differences in some of the deliverables between the US and EU, focused on integral DDCs with a primary mode of action for the drug substance(s). Developing a DDC using a systematic approach such as Quality by Design has consequences for the required supportive documentation, which increases with each subsequent phase of the product development and is mandatory to file.
Introduction
Pharmaceutical products such as tablets, suspensions, ointments, etc., are the traditional pharmaceutical dosage forms regulated in the US, usually by CDER, and in the EU by EMA or national health authorities. There are also medical devices, such as a syringe and an inhaler, as well as a specialized hospital bed or medical measuring equipment. Both have their specific regulations. What if you combine these two? In such a case, one speaks of drug-device-combination products (DDC), defined as the combination of a medicinal product or substance with a medical device. The different constituent parts of such a combination are regulated in the EU in the Medical Device Regulation, or MDR 2017/745, and similar requirements have been added to Directive 2001/83/EC. The quality requirements for regulatory submissions of a DDC product are presented in the fairly recent EMA guidance ‘Guideline on quality documentation for medicinal products when used with a medical device’ of July 2021 .
An example of such a product is intra-vaginal rings (IVR), where the active pharmaceutical ingredients, i.e., the medicinal part of the product, are dispersed in a slow-releasing matrix, and this matrix is seen as the device part of the product. More clarity on the differences between the EU and US regulatory aspects of this specific kind of DDC and the development thereof are presented hereafter.
DDC concepts
Integral versus non-integral devices
To understand which regulations apply, you first need to categorize the kind of product you are dealing with. In the EU, a distinction is made between integral DDCs and non-integral DDCs.
Integral DDCs are devices with the intent to administer a medicinal product, where they form a single integral product intended exclusively for use in the given combination and which is not reusable. Typically, these devices have measuring, metering, or delivery functions. Examples are pre-filled syringes, pre-filled inhalers, transdermal patches, and intra-vaginal rings (IVR). The term ‘single entity’ is used for integral DDCs in the US. However, the EU definition for this group of products has more detailed requirements, i.e., there is a partial overlap between the two product designations.
Non-integral DDCs are co-packaged and supplied along with the medicinal product, or where the product information refers to a specific device to be used in combination with the medicinal product and where the device is or can be obtained separately. Examples are reusable pens or inhalers for medicine-filled cartridges. These devices require a separate assessment with CE certification from notified bodies in the EU in contrast to the FDA assessment in the US.
Primary mode of action (PMOA)
Secondly, you must determine the primary mode of action (PMOA). The PMOA is defined as the primary goal of the drug-device combination. In the case of an IVR, the primary goal is the local or systemic administration of the substance(s), and the device part (the ring body) is the aid to administer the drug in the vagina, where it is absorbed, achieving either a local or systemic effect. Besides releasing the substance in a controlled manner, a suitable shape of the IVR combined with specific mechanical properties (e.g., flexibility) ensures that the ring remains in situ without causing irritation.
In the case of a non-integral DDC (e.g., an empty syringe or dry powder inhaler), the PMOA can be diverse, but in the two given examples, it is focused on consistent and/or precise dose delivery of the drug product. All safety and performance aspects of the device should be considered and addressed, including the product’s representation to the user (i.e., labeling).
EU
The IVR product is considered an integral DDC. Since the action of the substance(s) is principal and not ancillary to that of the device, the IVR product is regulated in the EU under Directive 2001/83/EC, i.e., the general drug guidance. When the substance(s) is used separately from the device part, it is considered a medicinal product of its own and regulated under that same directive.
If the substance’s action is not principal, the product is regulated under Regulation MDR 2017/745, the medical device guidance.
Figure 1 below depicts the process flow of the above-presented considerations to determine which regulation the DDC falls under.
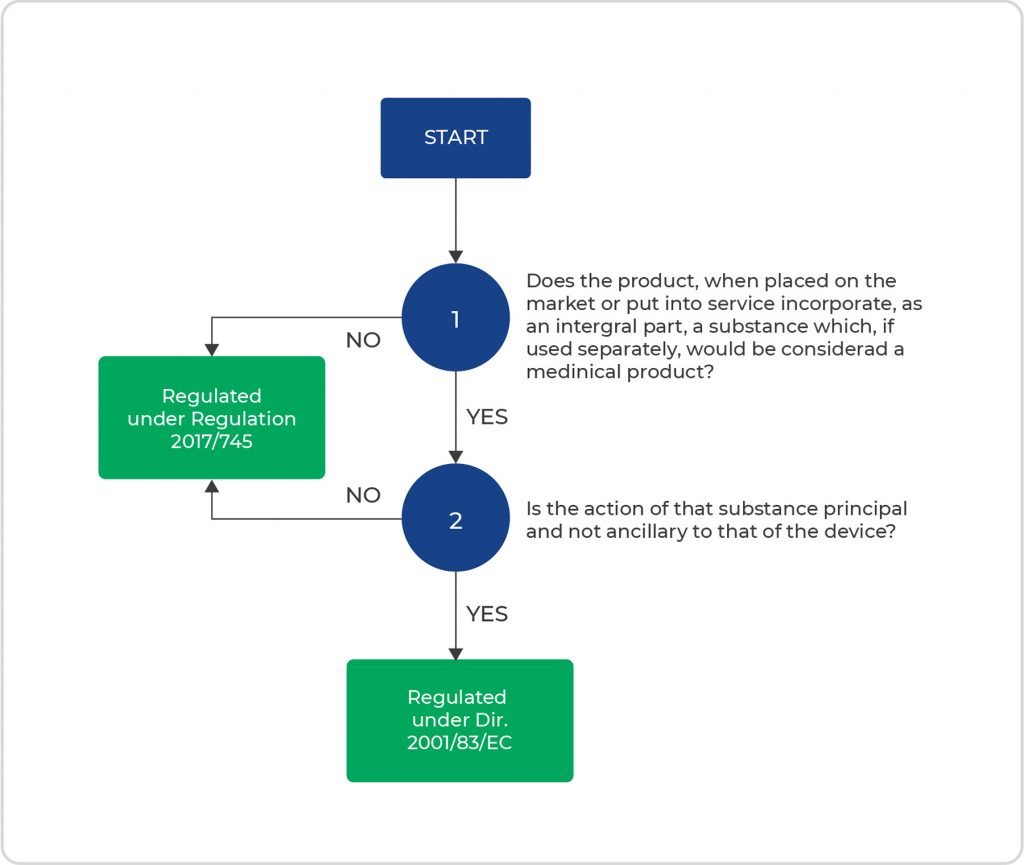
- Figure 1. Process flow of considerations to determine the proper regulation for DDCs.
US
The principles mentioned above also apply in the US. In the US, combination products are regulated in 21 CFR Part 4 of Subchapter A, with references to Subchapter C, parts 210 and 211, and Subchapter H, part 820. Additionally, there is guidance for industry documents on the cGMP requirements for combination products , derived from 21 CFR 820 for the device part and 21 CFR 210 and 211 for the drug part of the DDC. The Office of Combination Products (OCP) coordinates and assigns combination products to FDA’s medical product centers for review and oversees the postmarket activities.
Quality by design
The ICH definition of Quality by design (QbD) is “a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management.” It is meant to ensure that the intended performance of a final drug product is as expected, both in terms of safety and efficacy. To achieve this requires well-described objectives and proper risk management. The concepts behind quality by design were introduced in international guidelines (ICH) intended for the pharmaceutical industry between 2009 and 2012. ICH Q8, Q9, and Q10 are of great importance for drug products, describing the pharmaceutical development, quality risk management, and pharmaceutical quality system, respectively.
In the traditional quality-by-testing (QbT) approach, product quality and performance are ensured mainly by end-product testing, with a limited understanding of the process and critical process parameters. Regulatory bodies are therefore focusing on implementing quality by design (QbD), a science-based approach that improves process understanding by reducing process variation and enabling process-control strategies. The quality system expectations for medicinal products used with a medical device are mostly similar under EU and US regulations, but the deliverables differ, as described hereafter. Figure 2 presents the topics of interest and their content and presence throughout the different stages of clinical development, where the level of detail increases when moving toward registration. Quality by design does not stop after registration. It is continuously monitored utilizing a verification process that supports the annual product quality review during the entire product life cycle.
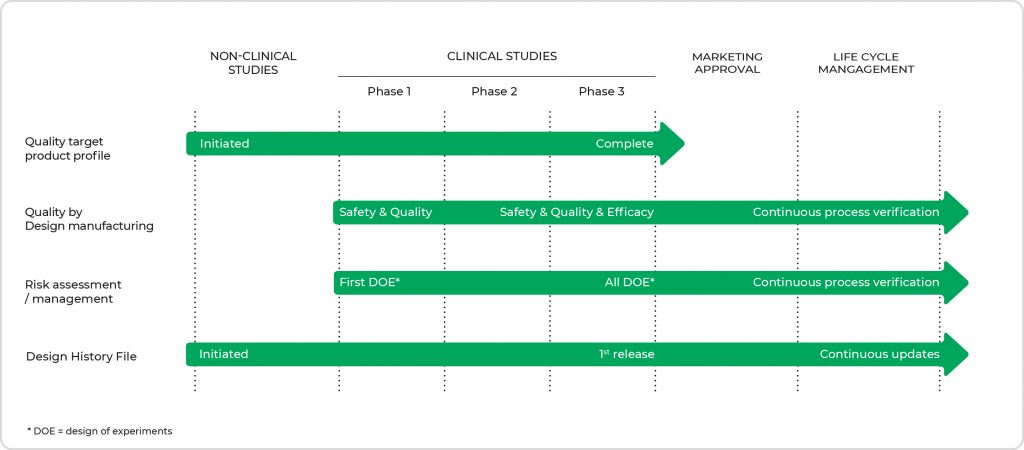
- Figure 2. Overview of the topics of interest with their main focus points throughout the different stages of clinical development. The level of detail increases when moving toward market approval. (DOE = design of experiments)
Quality Target Product Profile
Setting up a Quality Target Product Profile (QTPP) is an essential step in the QbD process as it is the basis for product development. Having a limited set of data at the start, it should be updated along the way with new insights and data obtained during development up to and including phase 3. It contains information on the expected product characteristics, such as intended use, product strength, and API release. Together with the design development plan, the design requirement specifications, the risk management plan and/or prior knowledge, critical quality attributes (CQA), and critical process parameters (CPP) are established, validated, and verified in the design process. While the FDA expects this document to be in the dossier, EMA does not address this specifically in its guidance. .
Risk Assessment & Management
Risk assessments are another necessary science-based process to identify which material attributes and process parameters can affect the product CQAs. They can be applied or assessed and updated at different stages in the product and process life cycle. Both FDA and EMA expect to see this information presented in the dossier. Annex I of ICH Q9 presents a general overview of some of the primary tools that might be used in quality risk management, where FMEA (Failure Mode Effects Analysis) and FMECA (Failure Mode, Effects, and Criticality Analysis) are perhaps the most used in the pharmaceutical industry. Both methods summarize the critical modes of failure, the factors causing these failures, and the likely effects of these failures. FMECA is an extension of FMEA, which incorporates an investigation of the severity of the consequences, their respective probabilities of occurrence, and their detectability.

Design Controls
The design control process during the development of a DDC is of great importance for both the US and the EU and is described in ICH Q8, for the US regulated under 21 CFR 820.30 with emphasis on the Design History File (DHF). The DHF is a collection of essential documents generated during the product life cycle. It describes the product design and, as such, a necessary set of documents for the FDA, which is subject to inspection during their audits. If the DHF does not conform to the requirements, the FDA will re-inspect until the conditions have been met. The DHF contains records or references to records that were necessary to demonstrate that the design was developed following the approved design plan and the device requirements. Although there is no specific requirement for a DHF in Europe, the MDR stipulates that ‘information to allow the design stages applied to the device to be understood’ should be present, which means that also, for the EU, one should compile a DHF. The DHF is a living document and should be maintained throughout the product’s lifecycle. All changes made to the product (e.g., engineering change orders, change of articles, bill of materials, or drawings) with a potential effect on the quality and efficacy before and after registration should be reflected in the DHF. Note that for non-integral DDCs, a crucial part of the medical device design and development process is to create a user and product requirement specification. The creation of this document takes place at the earliest stages of the process. It clearly defines the intended use, user needs, purpose, functionality, features, and behavior of the medical device idea and sets the basis for risk assessment, Design verification, and design validation.
Regulatory details throughout development
The level of regulatory detail differs per stage of development. The further into clinical development and closer to registration, the more detail must be provided in the various sections of the dossier. This is standard practice when developing regular drug products, and in the case of DDCs, this applies as well. However, for the device part in a DDC, the design history file starts right at the beginning of development as defined in the Design and Development plan. All trials, experiments, and changes need to be documented from the start of clinical development. Any changes in the design between the different clinical stages need to be bridged, if required, to ensure that whatever was established before the change still applies to the changed product. This is not different from the development of classical drug products, whereas differences in formulation that impact product efficacy should also be bridged between clinical trials. The same principle is applied in the EU and the US, although slight differences exist. The different submission formats partially cause these differences, whereas the US deals with a continuous update of the IND (investigational new drug application) and the annual reports to provide information on the development, and in the EU, only stand-alone IMPDs (investigational medicinal product dossiers) are provided.
Location of documentation in the dossier
Generally, drug product information for a DDC and its related engineering and manufacturing information should be located in the same dossier sections as would be provided for the drug product alone. For example, the CMC (chemistry, manufacturing, and controls) quality module should contain information on such device’s constituents in section 3.2.P.7. Supportive files for container closure device constituents should be presented in section 3.2.R. For other types of device constituent parts that do not have a logical location within 3.2.P, the information should be placed in 3.2.R. Quality information on the combination product as a whole (not the separate constituent parts) should be located in 3.2.P with appropriate hyperlinks to 3.2.R. As such, interrelationships between different sections of the dossier apply, with partial repeats from several sections to others, as presented in Figure 3.
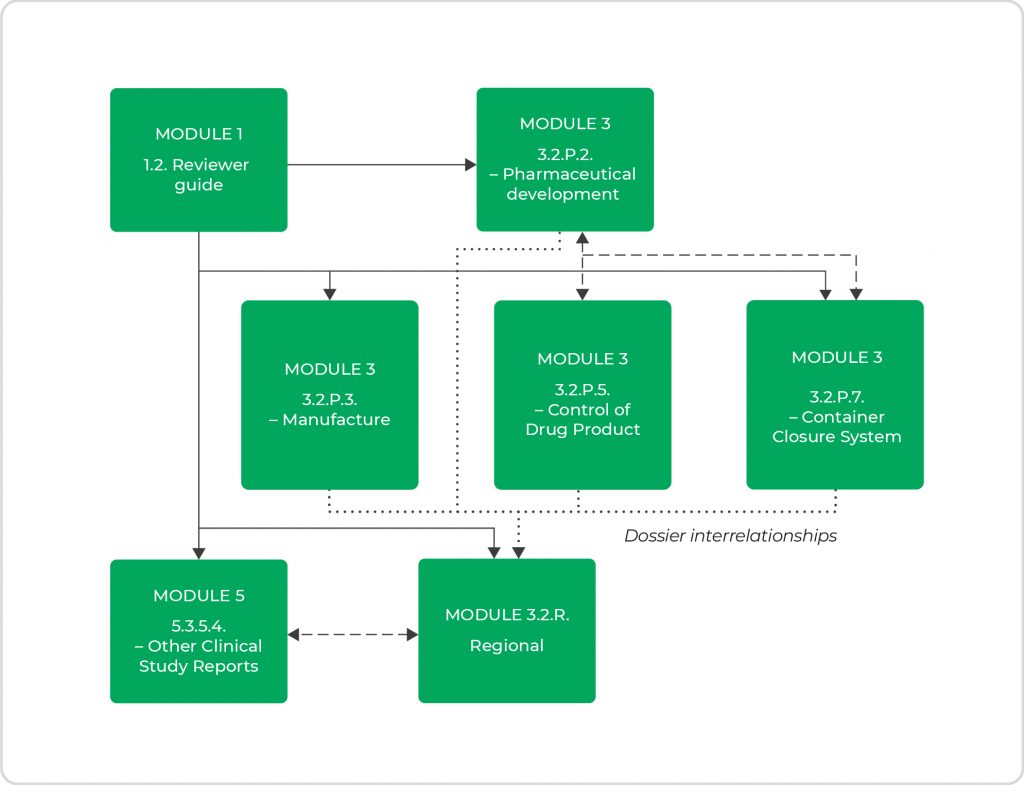
- Figure 3. Interrelationships of information between the different sections of the dossier.
US and EU guidance are generally quite specific about where to locate particular documents or information in the dossier. Yet, there are differences in what the different agencies expect to see in certain dossier sections. Module 3.2.R is such a module concerning, for example, the presence of particular parts or summaries of the Design History File for the US, while EU guidance does not stipulate that.
CONCLUSION
Not surprisingly, differences in guidance and expectations between agencies exist. Nevertheless, the guiding principles are the same, and, in the end, it all comes down to showing that you are in control of manufacturing a safe product of consistent quality that is efficacious for the patient. Understanding the differences in deliverables will support the different applications between territories and expedite agency assessments, potentially resulting in rapid market approval.
Acknowledgments
This document represents a personal perspective and does not confer any legal aspect or immunity to its user (Person or Legal Entity). The perspective is built on the study of the regulations and the personal understanding thereof, collegial discussion, and consensus, and, as such, not agreed upon by the Regulators (EMA, CMDh, or FDA).
REFERENCES
- 1] EMA/CHMP/QWP/BWP/259165/2019 – Guideline on quality documentation for medicinal products when used with a medical device, 22 July 2021.
- 2) Guidance for Industry and FDA Staff: Current Good Manufacturing Practice Requirements for Combination Products, January 2017.
- 3) Note for Guidance on Pharmaceutical Development (EMEA/CHMP/167068/2004), 22 June 2017
